New approach increases AlphaFold 2’s effectiveness for protein complexes
SciLifeLab researchers have managed to increase the effectiveness of the deep learning AI tool AlphaFold2 when predicting protein complexes. This was done by using optimized multiple sequence alignments and tweaking the original protocol. The researchers managed to obtain a 63 percent success rate in docking heterodimeric protein complexes compared to 45 percent when using the default protocol.
AlphaFold 2, a revolutionary AI deep learning tool developed by Google’s DeepMind project, has been praised by scientists all over the world and is said to have sparked a medical revolution within the Structural Biology field. AlphaFold 2’s main function is to predict the structure of single-chain proteins “in silico” and it does so much more accurately and faster than any previous software.
Predicting the structure of interacting protein chains is a fundamental step towards understanding their functions, but unfortunately, not even AlphaFold 2 is able to predict the structures of protein complexes completely. To increase the accuracy, scientists are now experimenting with new methodological approaches, where the AlphaFold 2 protocol is tweaked or combined with other techniques.
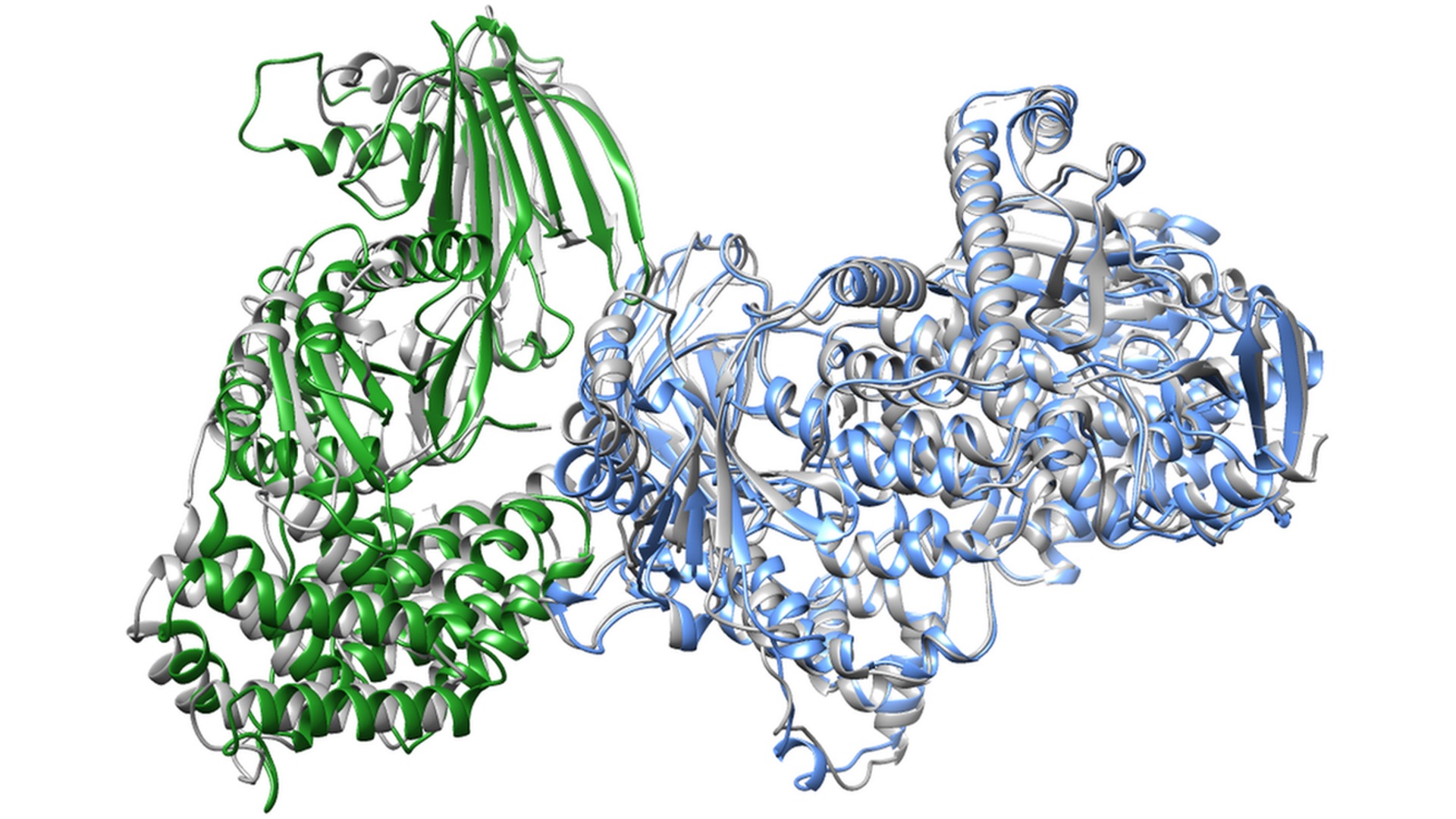
In a recent study, led by SciLieLab researcher Arne Elofsson, researchers wanted to investigate AlphaFold 2’s ability to predict the structure of heterodimeric protein structures, by combining the AlphaFold2 protocol with optimized multiple sequence alignments. By creating a simple function to predict the DockQ scores (pDockQ), the researchers could distinguish acceptable from incorrect models, and interacting from non-interacting proteins, with state-of-the-art accuracy. This has resulted in very accurate predictions of unknown protein complexes, as the prediction of phosphoinositide 3-kinase p110 gamma (7MEZ) shown below (experimental structure in grey and predicted in green and blue).
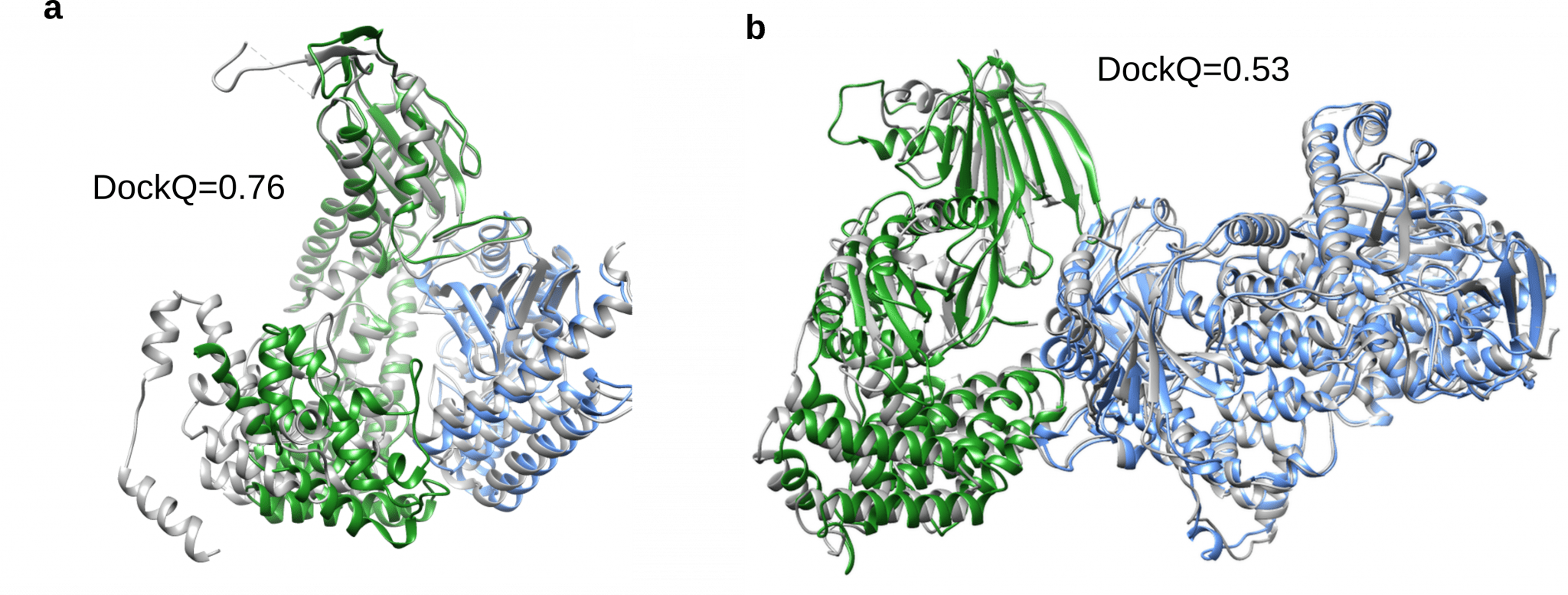
The results from the study, published in Nature Communications, shows a 63 percent success rate in docking heterodimeric protein complexes, by using the optimized multiple sequence alignments and scoring models using the number of contacts in the predicted interfaces. This can be compared with the 45 percent success rate of only using the default AlphaFold 2 procedure.
In summary, the researchers have, for the first time, demonstrated that a few tweaks to the AlphaFold 2 protocol can provide a significant step towards identifying the structural basis for many protein interactions in a cell.