

Arne Elofsson
Research interests
Life science has undergone a revolution in the last decades, from a data-poor to an extremely data-rich scientific field. However, the increase in novel data is not evenly distributed. Some types of data, such as individual functional classifications of proteins, have only increased slightly, while other types, such as genomic sequence data, have undergone an exponential increase. Simultaneously, deep-learning (or AI) methods have advanced during the last decade. Therefore, how to best use the deep-learning (or AI) method in life science is essential for efficiently utilising all the large-scale data generated by high-throughput molecular life-science techniques. Although this has been claimed for the last decade – the progress has been surprisingly small.
One of the most impressive signs of progress in AI-based life science was reported in December 2020 at the CASP14 conference. There DeepMind showed that their method, AlphaFold2, could predict the structure of basically all proteins (for a summary see: https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology). In short their method builds on earlier work to predict contacts (or distances) between residues in a protein (as we do in our PconsC1-4 methods). Already in CASP13 DeepMind showed progress in this field. Still, this progress was only a small step ahead of the competition, and today several alternative methods exist that perform as well as their earlier method. In contrast, AlphaFold2 was better than all other methods for 90% of the models. The method also provided a completely novel architecture with an end-to-end learning approach. This shows that we can combine large-scale biotechnological data (sequences) with deep learning (AI) to obtain real biological knowledge (protein structures).
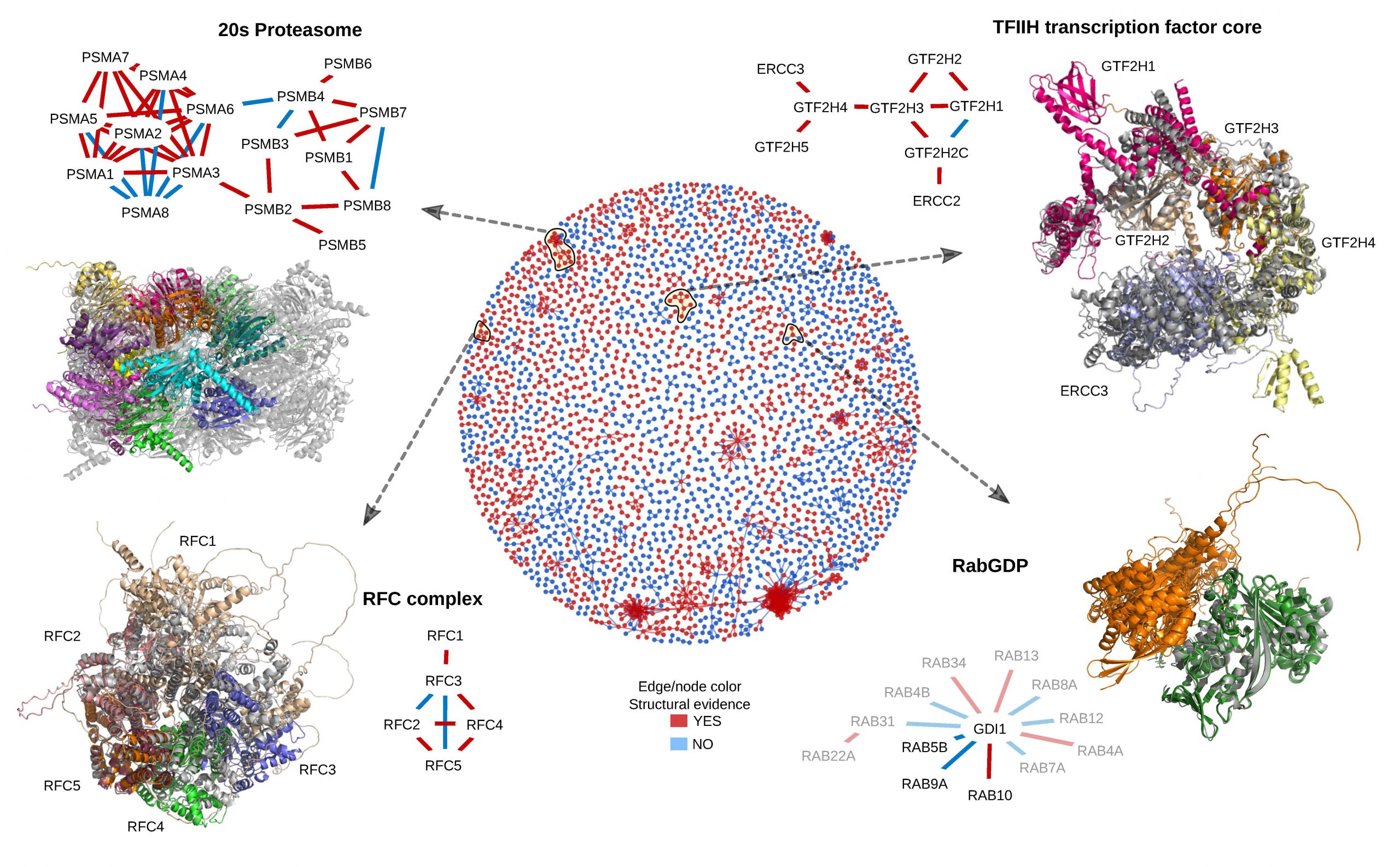
However, proteins do not act alone. They function by interacting with other proteins and other molecules. Protein interaction can vary in nature from stable interaction present in small and large protein complexes to transient interactions often used for regulation. Prediction of protein interactions has been a larger challenge than the prediction of the structure of individual proteins and progress has been limited in the last decade. Many different techniques have been developed, but they can be divided into three categories. We are developing and applying novel methodologies for protein-protein structure predictions. We use a fold-and-dock protocol based on alphafold, where interacting proteins are folded and docked simultaneously using information from predicted residue-residues interactions. This method has enabled us to predict the structure of many interacting proteins and even large complexes.
Group members
Principal Investigator: Arne Elofsson
Sarah Narrowe Danielsson
Samuel Fromm
Petras Kundrotas
Gabriele Pozzati
Aditi Shenoy
Christopher Sprague
Wensi Zhu