

Mass spectrometry (MS)-based methods, capable of proteome-wide analysis of cells and lysates, hold immense potential to unravel how various compounds or molecules function by measuring their effects on the proteome. For first-in-class drugs, or those identified through phenotype-based approaches—such as after phenotypic screening or cell phenotype assays—it is crucial to ensure accurate target deconvolution (TD) of candidate drugs. This helps elucidate early mechanisms of action (MoA) and triggered functional cascades in cells, which are vital for the effective application of candidate drugs, repurposed molecules, novel therapies, or drug implementation strategies.
In target-based approaches, such as in studies of follower drugs, Hydrogen/Deuterium Exchange (HDX)-MS is particularly useful. It facilitates binding site mapping and the characterization of protein interactions with small molecules, peptides, or other ligands—including proteins involved in epitope mapping in antibody-antigen interactions. HDX-MS also enables monitoring of conformational changes in proteins to study the effects of interactions, mutations, protein misfolding, and biosimilar characterization, among other applications.
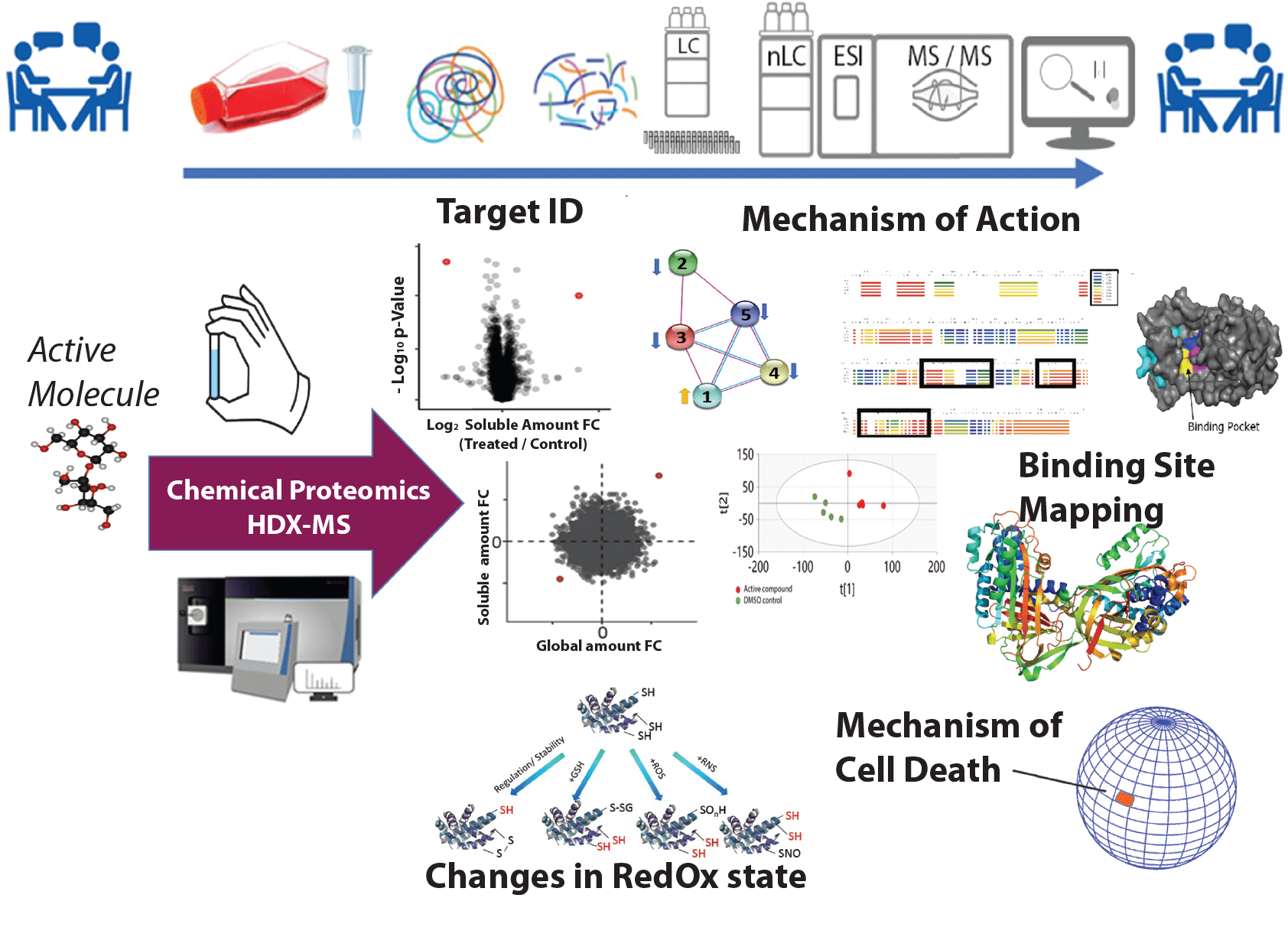
Chemical Proteomics is the unique national unit of SciLifeLab, the unique node of BioMS for chemoproteomics support. In both these infrastructures, and one of the EU-OpenScreen Chemoproteomics partner sites. Our interest is to adopt and develop the most efficient methods in chemoproteomics, for providing research support and expertise, within and outside Sweden, for elucidating the mechanism of action (MoA), target engagement and target deconvolution (TD) of any drugs or compounds of interest in research projects. We also support target-based approaches, as in the case of studies on follower drugs, providing by HDX-MS: binding site mapping and characterization of protein interactions with small molecules, peptides, or any other ligand, including proteins, such as for the cases of epitope mapping in antibody-antigen interactions; monitoring conformational changes on proteins to study effects of interactions, mutations, protein misfolding and for biosimilar characterization, etc.
Chemical Proteomics offers a complete support pipeline to research projects, from the initial project’s discussion and definition of the experimental design tailored to the research questions, up to full data analysis, visualization and data interpretation. The workflows are coordinated from the initial reception of samples, for example cells to be cultured at our laboratories and compounds for their treatment, with the aim to accurately reproduce the conditions used by the requesting PI´s for the existing research data or compound testing.
Our recent MS-based and proteome-wide approached also resolved relevant bottlenecks of the proteomics methods for TD and MoA characterization. In particular our Proteome Integral Solubility Alteration assay (“PISA”) nowadays a well-recognized trademark by the scientific community, adopted as the leading methodology for TD by the most relevant proteomics laboratories in the world, and our request of support using PISA is dramatically increasing. The early changes of solubility of proteins, typically observed within 60 minutes of treatment in living cells, provide insights into compound-specific interactions and cellular events involving key MoA protein factors. The development and optimized application of current technologies—and their ongoing advancements—enable experimental designs requiring minimal sample amounts, making them suitable for precision medicine and medicinal chemistry strategies.
Given that each methodology for deciphering the MoA comes with technical limitations at different stages, it is essential to design tailored analyses with a robust experimental plan, possibly integrating orthogonal proteomics approaches to measure proteome-level changes across different dimensions of proteome-wide analyses. We tailor the experimental design on the projects´ needs offering exploitation and integration of orthogonal methods, also improved and/or developed in-house. Among them: time-resolved expression proteomics; FITExP or expression proteomics analysis combined with the ProTargetMiner database for anticancer molecules; high throughput RedOx Proteomics; affinity/activity-chemoproteomics; analysis of post-translational modifications; FT-IsoR-MS for isotope composition of proteins; H/D exchange MS (HDX-MS).
This integration must also balance high-throughput capabilities with sustainable costs and timelines. Without this, phenotypic screenings often fail to reveal the molecular deconvolution of cause-and-effect alterations at the proteome level, which is essential for drug discovery and development.
Group Members:
Hassan Gharibi, PhD
Olga Lytovchenko, Ph.D.
Susanna Lundström, Ph.D.
Xuepei Zhang, Ph.D.
Zhaowei Meng