

National Genomics Infrastructure
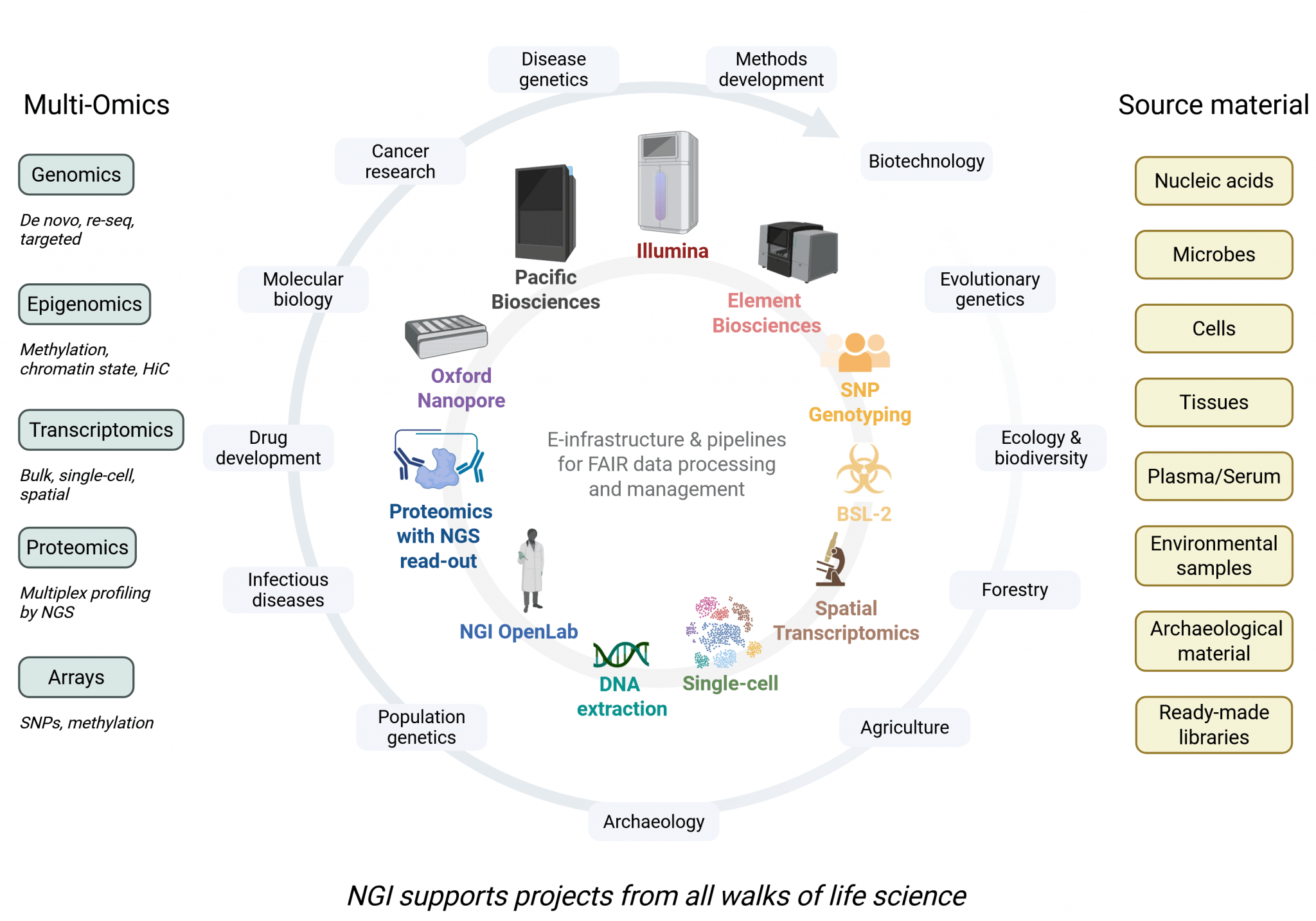
The National Genomics Infrastructure (NGI) at SciLifeLab is a leading national research infrastructure for molecular biosciences, supporting a wide range of life science disciplines and research fields, including health, environmental research, and technology development.
As part of SciLifeLab’s Genomics Platform, NGI provides access to various advanced technologies for library preparation, sequencing, genotyping, and bioinformatics support. By providing these state-of-the-art resources, NGI aims to facilitate and enable research at all scales – from SNPs to whole genomes, from single cells to tissues – while ensuring high-quality data generation and analysis.
The platform operates through nodes in Stockholm (NGI Stockholm) and in Uppsala (NGI Uppsala; SNP&SEQ Technology Platform and Uppsala Genome Center).
Our Mission
- Enable world-class research and industrial development in genomics.
- Provide access to both established and cutting-edge genomics technology.
- Assist users in project design, data generation, analysis and management of data.
- Support genomics research in all subject areas.
- Support scientists, core facilities and industry in all parts of Sweden.
- Support data-driven science according to FAIR principles.
- Attract and retain highly skilled staff.
Contact Us
For any inquiries, including questions about NGI’s services, please contact us at support@ngisweden.se or visit our website: ngisweden.scilifelab.se